 を押して省略できます。
を押して省略できます。はじめに
生物の遺伝情報を保持しているDNAは、複製時のDNApolymeraseのエラーに加えて、環境中の放射線や紫外線、またはアルキル化剤等の化学物質、生体内における活性酸素等の代謝産物により損傷を受ける。これらのエラーや損傷が正しく修復されなければ突然変異を誘発し、これが癌や老化の原因となる。DNA損傷部位には修復機構が働き、その一つとして塩基除去修復がある。この時APsite(apurinic/apyrimidinicsite)と呼ばれる塩基除去部位が出現する。つまりAPsiteの検出はDNA損傷部位を測定し得る有効な方法となる。-Nucleostain-DNADamageQuantificationKit-APSiteCounting-は、APsiteと特異的に結合するARP(N’-Aminooxymethylcarbonyl-hydrazino-D-biotin)を用いてDNAをビオチン化し、96穴マイクロプレートに固相化して試料DNA中のAPsiteを簡便に定量できるキットである。本キットには、精製牛胸腺DNAから調製したAPsite数の既定されたARPStandardDNAが含まれており、これを用いて検量線を作成することにより試料DNAのAPsite数を定量することができる。
キット内容
ARP-DNA Standard Solution
-
0 ARP-DNA Standard Soln.※
(0 ARP/100,000 bp)250 µl x 1 2.5 ARP-DNA Standard Soln.※
(2.5 ARP/100,000 bp)250 µl x 1 5 ARP-DNA Standard Soln.※
(5 ARP/100,000 bp)250 µl x 1 10 ARP-DNA Standard Soln.※
(10 ARP/100,000 bp)250 µl x 1 20 ARP-DNA Standard Soln.※
(20 ARP/100,000 bp)250 µl x 1 40 ARP-DNA Standard Soln.※
(40 ARP/100,000 bp)250 µl x 1
-
ARP Solution (10 mmol/l ARP) 250 µl x 1 DNA Binding Solution 10 ml x 1 Substrate Solution 10 ml x 1 TE Buffer 40 ml x 1 HRP-Streptavidin 25 µl x 1 Washing Buffer (powder for 1 L) 1 packet Filtration Tube 20 tubes 96-well Microplate/ U bottom 1 plate Manual 1 booklet - ARP-DNA Standard Solutions: 0.5 µg DNA / ml
キット以外に必要なもの
-
- 10 μl, 200 μl, 1 ml マイクロピペッター ( 可変式 )
- 0.5 ml, 1.5 ml 遠心チューブ
- 200 μl 8 連マイクロピペッター(可変式)
- 遠心機
-
- インキュベーター (37℃ )
- DNA 精製キット ※
- マイクロプレートリーダー
- ペーパータオル
- 本キットは混在する RNA およびタンパクにより、DNA の AP site 数に検出測定誤差を生じる可能性があるため RNase A 処理後、RNA とタンパク質を除去する必要があります。DNA 精製は、市販のキットをご利用ください。
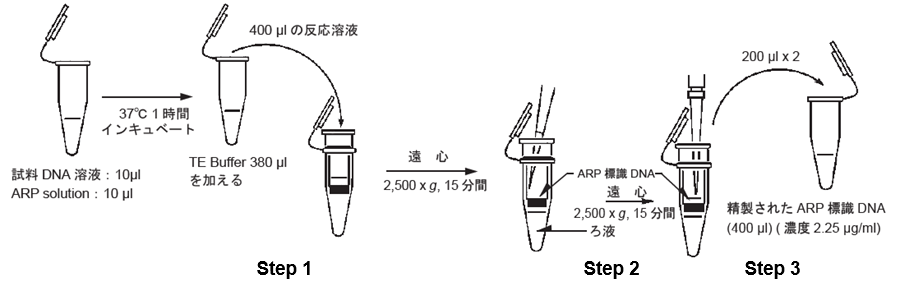
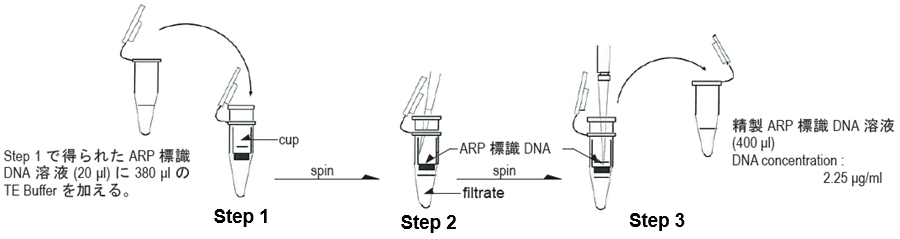
- Step 1
cupを100 μl のTE Buffer で2回洗浄する。400 μl のARP標識DNA溶液をFiltration Tubeに入れ2,500 x g で15分間遠心する。
- Step 2
ろ液を捨て、再び400 μl のTE Buffer をFiltration Tubeに加えて溶解後、2,500 x g で15 分間遠心する。
- Step 3
200 μl のTE Bufferを加えて、フィルター上に付着したDNAを溶解させ溶液を1.5 ml 遠心チューブに移す。
再び、200 μl のTE Bufferを加えてフィルター上に残ったDNAを溶解させ、同様に1.5 ml 遠心チューブに移す。
図 1 試料 DNA の ARP 標識
操作方法
1. 試料 DNA の ARP 標識 ( 図 1 を参照 )
- 精製した試料 DNA を TE Buffer※1 に溶解させ、溶液の吸光度 (260 nm) を測定する。吸光度から濃度を算出して (DNA 濃度は 50 μg/ml の時の吸光度を 1.0 として算出 )100 μg/ml の濃度になるように TE Buffer※1 で希釈調製する。
- 試料 DNA 溶液 10 μl と ARP Solution 10 μl を 0.5 ml 遠心チューブ中で混合する。
- 37°Cで 1 時間インキュベートする。その後、380 μl の TE Bufferを添加する。
- まず、Filtration Tube の cup を 100 μl の TE Buffer で 2 回洗浄する。操作 3. の溶液を Filtration Tube に入れる。
- 遠心分離 (2,500 x g、15 分間 ) 後、ろ液を捨てる ※2。
- 操作 5. の Filtration Tube に 400 μl の TE Buffer を加え、フィルター上に付着した ARP 標識 DNA をピペッティン グにより溶解させる(注意事項 3 参照)。
- 再び遠心分離 (2,500 x g、15 分間 ) 後、ろ液を捨てる ※2。
- 操作 7. の Filtration Tube に 200 μl の TE Buffer を加え、フィルター上に付着した ARP 標識 DNA をピペッティン グにより溶解させる(注意事項 3 参照)。
- 溶解させた DNA 溶液を 1.5 ml 遠心チューブに移す。
- さらに 200 μl の TE Buffer を 操作 8. の Filtration Tube に加え、操作 8. の操作をもう一度繰り返し、得られた ARP標識DNA 溶液を 操作 8. の DNA溶液と合わせる ※3。
- 保存の必要がある場合、1.5 ml 遠心チューブに移した ARP 標識 DNA 溶液を 0 〜 5°Cで保存する。
- 本キットには精製した試料 DNA の溶解または、希釈に使用する TE Buffer は含まれていないので、次のように調製する。
TE Buffer(500 ml):滅菌水 500 ml に Tris を 606 mg(10 mmol/l)、3NA を 206 mg(1 mmol/l) 溶解し、6 mol/l の HCl で pH を 7.5 に調整後、オートクレーブ滅菌する。 - 溶液がメンブラン上に残っている場合は、更に 2,500 x g、5 分間遠心分離する。
- Filtration Tube を使用した場合の DNA 回収率は 90% である。よって、溶液中の ARP 標識 DNA 濃度は 2.25 μg/ml となる。
2. AP site 数の検出
- 操作方法 −1 で調製した ARP 標識 DNA 90 μl を 1.5 ml 遠心チューブに入れ、TE Buffer 310 μl を加える。
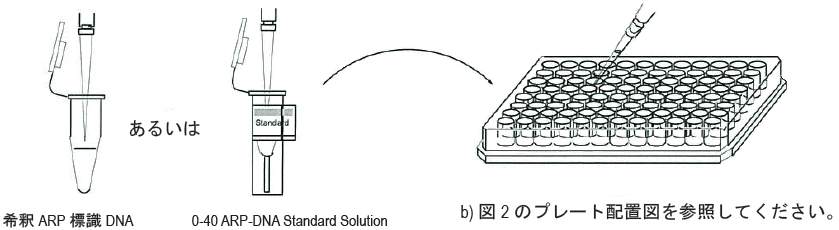
- 各 ARP-DNA Standard Solution を 1 ウェル当たり 60 μl ずつ 3 ウェルに添加する(「プレートレイアウト」参照)。
- 操作 1.で調製した ARP 標識 DNA を 1 ウェル当たり 60 μl ずつ 3 ウェルに添加する。
- DNA Binding Solution 100 μlをDNAの入ったウェルに添加し数回ピペッティングした後、室温で一夜放置する。
- 添付の Washing Buffer 粉末をイオン交換水 1 L に溶解して洗浄用 PBST を調製する。HRP-Streptavidin を洗浄用 PBST で 4,000 倍に希釈し、希釈 HRP-Streptavidin を調製する(例 HRP-Streptavidin 10 μl + PBST 40 ml)。
- ウェル中の溶液を吸引などにより除去し、洗浄用 PBST 250 μl でプレートを洗浄する。再びウェル中の溶液を 吸引などにより除去し、ペーパータオルの上でプレートを叩いてウェル中の残存溶液を完全に除く。この操作を 5 回繰り返す。
- 希釈 HRP-Streptavidin 150 μl を各ウェルに添加し、37°C、1 時間インキュベートする。
- 操作 6. と同様にして、洗浄用 PBST でプレートを 5 回洗浄する。
- Substrate Solution 100 μl を各ウェルに添加し、37°C、1 時間インキュベートする。
- マイクロプレートリーダーにより、吸光度を測定する。630 〜 670 nm の波長フィルターが使用可能である。
- DNA 中の AP site 数を検量線から検出する(図 2)。
-
プレートレイアウト
RP-DNA Standard Solution あるいは ARP 標識 DNA(60 μl) と DNA Binding Solution(100 μl)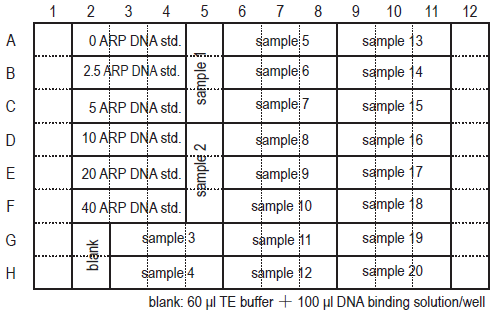
-
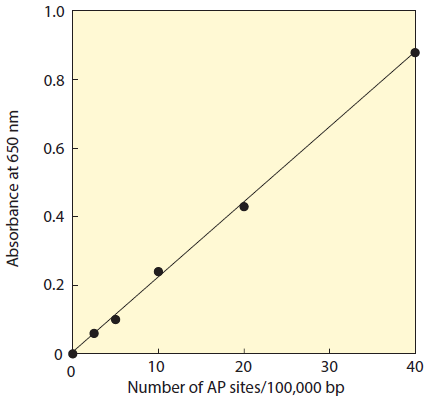
図2 ARP-DNA Standard Solutionを用いて作成した検量線例
注意事項
- キットは0~5°Cで保存し、凍結させないで下さい。
- 試料DNAのAP siteは一般に不安定ですので、測定対象DNAを単離精製後は直ちにARP標識を行ってください。ARP標識DNA溶液は0~5°Cで1年安定です。
- Filtration Tubeでの遠心分離後は直ちにTE Bufferを添加しDNAを溶解してください。長時間DNAをメンブラン上で放置しておきますと、DNAのメンブランへの吸着が起こり回収率にバラツキを生じる可能性があります。
- γ線滅菌のチューブはDNAの吸着を生じる可能性がありますので、チューブを使用の際は非滅菌チューブを必要に応じてオートクレーブ滅菌して使用することを推奨します。
- 630~670 nm のフィルターを持ち合わせていない場合は、発色反応後、各ウェルより50 μl 抜き取り新しいプレートに移します。同量の1mol/l 硫酸を添加後、450 nm の吸光度で測定することができます。硫酸添加後、速やかに測定してください。
- ウェル洗浄後、残存する溶液により測定誤差を生じることがありますので完全に除いてください。
トラブルシューティング
A.発色しないあるいは発色が極端に少ない
- 試料DNAのプレートへの固定にDNA Binding Solutionを使用しましたか。不十分な量のDNA Binding Solutionを使用するとDNAがプレートにうまく固定されません。
- 1/4,000 に希釈したHRP-Streptavidin溶液を使用しましたか。
- Substrate Solutionを加えましたか。
- 正しい波長で測定していますか。
- 希釈HRP-Streptavidin溶液は使用直前に調製しましたか。この溶液は長時間の保存には適しません。
- HRP-Streptavidin溶液は適温(0~5°C)で保存されましたか。室温あるいは冷凍状態での保存は酵素活性を著しく低下させます。
B.試料DNAが発色しない
- 用意した試料DNAの濃度は100 μg/ml でしたか。
- 試料DNAは精製しましたか。
- 試料DNA溶液にARP Solutionを加えましたか。
- ARP標識後、Filtration Tubeを用いて精製しましたか。
- 試料DNA中のAP site数が検出限界である1/100,000 bpより少なくありませんでしたか。
C.全てのウェルの発色が強すぎる
- HRP-Streptavidin 溶液は、洗浄用PBSTで1/4,000 に希釈しましたか。高濃度のHRP-Streptavidin溶液は高バックグラウンドの原因になります。
- 洗浄用PBSTで各ウェルを5回ずつ洗浄しましたか。
D.検量線が直線にならない/ばらつきが大きい
- 各ARP-DNA Standard Soln.を同量ずつ使用しましたか。
- 洗浄用PBSTで各ウェルを5回ずつ洗浄しましたか。
- 洗浄時に洗浄用PBSTを完全に除きましたか。洗浄用PBSTを除いた後、完全に水気を取るためにペーパータオルの上でプレートを逆さにして数回叩いてください。
- 洗浄用PBSTはキット付属のWashing Buffer 粉末を純水 1L に溶解したものを使用して下さい。高濃度あるいは低濃度の洗浄用PBSTを使用すると大きな誤差を生じることがあります。
- DNAのプレートへの固定のため、プレートを室温で一晩放置しましたか。DNAを十分固定するためには少なくとも4時間のインキュベートが必要です。
- ARP標識後の精製はFiltration Tubeを用いた方法で行いましたか。ARPのコンタミネーションは測定に重大な問題を引き起こす可能性があります。
- 1 サンプルに1つのFiltration Tubeを使いましたか。
E.試料DNAの測定で測定可能レンジを超える
- 試料DNAを0 ARP-DNA Standard Solutionで希釈して測定して下さい。測定値に希釈倍率を掛けた値が試料DNAのAP site数となります。
- Filtration Tubeで精製した後、DNA溶液をTE Bufferで希釈しましたか。
- 取り出した試料DNAは十分精製されていますか。
参考文献
- T. Lindahl and B. Nyberg, Biochemistry, 1972, 11, 3610.
- J. Nakamura, V. E. Walker, P. B. Upton, S. Y. Chiang , Y. W. Kow and J. A. Swenberg, Cancer Res., 1998, 58, 222.
- K. Kubo, H. Ide, S. S. Wallace and Y. W. Kow, Biochemistry, 1992, 31, 3703.
- H. Ide, K. Akamatsu, Y. Kimura, K. Michiue, K. Makino, A. Asaeda, Y. Takamori and K. Kubo, Biochemistry, 1993, 32, 8276.
- A. Asaeda, H. Ide, K. Tano, Y. Takamori and K. Kubo, Nucleosides & Nucleotides, 1998, 17, 503.
General Protocol at a Glance
このGeneral Protocol を使用する前に取扱説明書を良くお読みください。
Day 1
|
Step 1 |
10 μl a) の試料DNA 溶液と10 μl a) のARP Solution を混合し37°Cで1 時間インキュベートする。 |
 |
|
|
Step 2 |
ARP標識DNAをFiltration Tubeで精製する。 |
- Step 1
まず、cupを100 μlのTE Bufferで2回洗浄する。400 μlのARPラベル化DNA溶液をFiltration Tubeに入れ2500 x g で15分遠心する。
- Step 2
濾液を捨て、400 μlのTE Bufferを加えARP標識DNAを溶解後、再び2500 x g で15分遠心する。
- Step 3
200 μlのTE Bufferを加えフィルター上に付着したARP標識DNAを溶解する。ARP標識DNA溶液を1.5 mlチューブに移す。この操作をもう一度繰り返す。
|
Step 3 |
90 μlのARP標識DNA溶液をチューブに取り、310 μlのTE Bufferで希釈する。60 μlの希釈溶液、あるいはARP-DNA Standard Solutionをウェルに加える b)。 |
|
Step 4 |
100 μlのDNA Binding Solutionをそれぞれのウェルに加え、数回ピペッティングしたのち室温で一晩放置する。 |
 |
 |
Day 2
|
Step 5 |
ウェル中の溶液を除去し、洗浄用PBST 250 μlでプレートを5回洗浄する。この時ウェル中の残存溶液を完全に除くため、ペーパータオルの上でプレートを逆さにしてたたくと良い。 |

|
Step 6 |
希釈 HRP-Streptavidin c) 150 μl を各ウェルに添加し 37°Cで 1 時間インキュベートする。 |
|
|
|
Step 7 |
Step 5 と同様にして、洗浄用PBST でプレートを5 回洗浄する。 |
|
Step 8 |
100 μl のSubstrate Solution を各ウェルに添加し37℃で1 時間インキュベートする。 |
|
|
|
Step 9 |
マイクロプレートリーダーで650 nm d) の吸光度を測定する。DNA 中のAP site 数を検量線から算出する。 |
 |
d) もし650 nm のフィルターを持ち合わせていない場合、発色後各ウェルより50 μl づつ抜き取り新しいプレートに移し、50 μl の1 mol/l 硫酸を添加し450 nm の吸光度を測定することができます。 |
よくある質問/参考文献



DK02: -Nucleostain- DNA Damage Quantification Kit -AP Site Counting-
Revised Jan., 10, 2024